美国FDA 于2019 年11 月发布了《透皮和局部递药系统– 产品研发和质量考量》指南草案,以帮助推进经透皮给药或局部传输系统(topical delivery systems TDS)新药和仿制药的开发。TDS产品应用于患者的皮肤,顾名思义,透皮给药系统是指药物以一定速率通过完整的皮肤表面,经毛细血管吸收进入体循环而产生疗效的一类给药系统,常见剂型包括凝胶贴膏(原巴布膏剂)、透皮贴剂、液体类制剂(洗剂、搽剂、酊剂等)、软膏和乳膏剂、凝胶剂、涂膜剂等[1-2];皮肤局部给药系统主要作用于皮肤局部或者局部皮肤下的各层组织,主要在局部发挥作用,常见制剂种类包括贴剂、软膏剂、乳膏剂、糊剂、凝胶剂、喷雾剂、涂剂、气雾剂、泡沫剂和散剂等[2-3]。
《中国药典》与《美国药典》对于贴剂与贴膏剂的分类和归属略有不同。《中国药典》2020 版中规定贴剂包括产生全身作用的透皮贴剂和局部作用的贴剂,贴膏剂包括可产生全身性或局部作用的凝胶贴膏与橡胶贴膏[4]。《美国药典》把使药物均匀释放或靶向到身体的贴剂、贴膏剂等制剂则都归属于递送系统,药物通过皮肤输送到全身循环而发挥作用的称为透皮给药系统[5]。
经过半个世纪的发展,透皮给药系统以其能够克服口服给药带来的肝脏首过消除、服药量大、较难保持血药浓度、对胃黏膜的刺激等缺点的优势,成为国内产品研发或引进的新热点[6-8]。国内外获批上市的透皮给药系统以透皮贴剂为主,主要用于戒烟、缓解疼痛、骨质疏松症、避孕、晕动病、心绞痛和心脏疾病等[8]。另外,凝胶贴膏具有载药量大,给药剂量准确,吸收面积小,血药浓度稳定,应用方便舒适等优点[9],在临床已应用于外伤所致肿胀及疼痛、关节炎、肌肉痛、风湿痛和神经痛等疾病。
本文根据美国食品药品监督管理局(FDA)相关指南,对贴剂与贴膏剂仿制药的生物等效性(bioequivalence, BE)研究的方案设计、试验操作中的关键点进行分析与总结,以期为其BE 研究的规范性、科学性提供参考。
国内贴剂与贴膏剂BE 研究相关法规概况
我国已发布的《化学仿制药参比制剂目录》中,对贴剂与贴膏剂的参比制剂进行了明确规定,《化学仿制药参比制剂目录》(第五十二批)共公布13 个有效成分的贴剂与贴膏剂参比制剂。透皮贴剂包括芬太尼、丁丙诺啡、利斯的明、奥布西宁;凝胶贴膏包括吲哚美辛、氟比洛芬、利多卡因、酮洛芬;贴剂包括妥洛特罗、洛索洛芬钠、双氯芬酸钠、酮洛芬、氟比洛芬。
国家药品监督管理局药品审评中心2021 年3月发布的《皮肤外用化学仿制药研究技术指导原则(试行)》中BE 评价部分明确对于局部作用的药物可能会导致全身暴露,且存在一定的全身不良反应风险时(如皮质类固醇激素类产品等),应首先开展仿制药与参比制剂之间人体药代动力学(PK)对比试验,比较两者在系统暴露方面的一致性,以支持其系统吸收相关的安全性评价[12],但是尚未对透皮贴剂、贴膏剂以及局部作用贴剂的BE 研究提供具体指导。
分析近5 年药物临床试验登记与信息公示平台的备案信息显示,透皮贴剂和凝胶贴膏BE 试验共有31 个备案,其中15 个备案信息在开展以PK 为终点的BE 研究(PK-BE)的同时观察了黏附性,5 个备案信息为PK-BE、黏附性和皮肤刺激性合并开展,未见单独的皮肤刺激和致敏性试验备案。总体来看,透皮贴剂与贴膏剂仿制药的黏附性研究和刺激性与致敏性研究并未受到重视。已备案的信息中黏附性研究的受试者例数,评估标准等是否合理仍需关注,而PK-BE 试验与皮肤刺激性研究合并开展,单次给药与给药时间是否满足观察制剂的长期或多次给药后的皮肤刺激性仍需规范。
FDA 贴剂与贴膏剂BE 研究概况
FDA 贴剂与贴膏剂《特定药物的生物等效性指导原则》概况
截止至2022 年2 月,FDA 已发布28 个贴剂和贴膏剂,涉及22 个产品的《特定药物生物等效性指导原则》,指南涉及22 个透皮贴剂、1 个透皮系统(乙炔雌二醇/ 左炔诺孕酮透皮系统)、5个局部作用贴剂(辣椒素贴剂、双氯芬酸依泊胺贴剂、薄荷醇/ 水杨酸甲酯贴膏、1.8% 利多卡因凝胶贴膏、5% 利多卡因凝胶贴膏),具体药品的通用名及商品名见表1。其中,睾酮、雌二醇、雌二醇/ 醋酸炔诺酮、硝酸甘油、可乐定、东莨菪碱透皮贴剂,利多卡因凝胶贴膏7 个产品在我国已仿制上市,罗替高汀透皮贴剂与薄荷醇/ 水杨酸甲酯贴膏的参比制剂NEUPRO、SALONPAS已在我国进口上市。
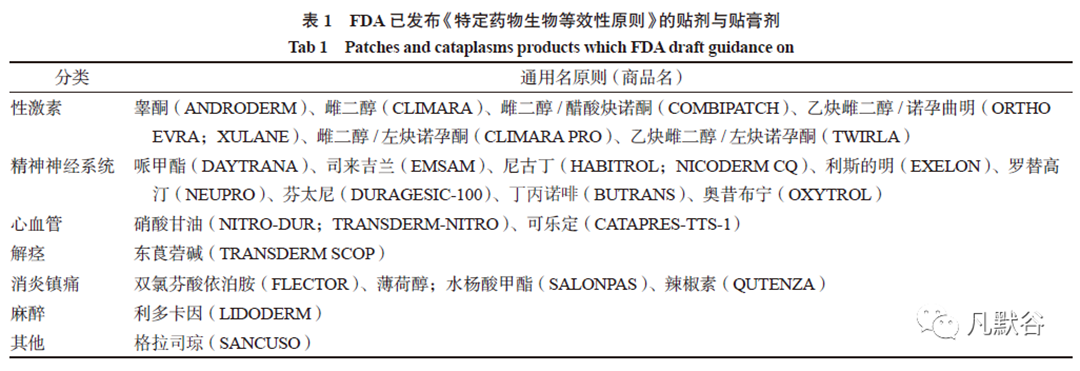
FDA 贴剂与贴膏剂的BE 评估方法
FDA 在《特定药物的生物等效性指导原则》(BE 指南)中,要求贴剂与贴膏剂需开展:①PK-BE 研究和/ 或临床终点BE 研究;② 黏附性研究;③皮肤刺激与致敏性研究(简称I/S 研究)。本文将分别详述FDA 贴剂与贴膏剂BE 评估方法的设计及关键点。
从FDA 已发布的28 个贴剂与贴膏剂的BE指南看出,透皮制剂和贴膏剂均采用PK-BE 方法,局部作用贴剂双氯芬酸依泊胺与辣椒素采用的是以临床终点生物等效性研究,此外,双氯芬酸依泊胺贴因在应用中可能发生严重的心血管血栓或胃肠道的不良反应,还采用了PK-BE 推测和衡量可能发生的不良反应。贴剂的黏附性、皮肤刺激性及致敏性均影响制剂的使用,进一步影响制剂中药物的吸收和制剂的安全性[10-11]。根据BE指南,贴剂与贴膏均需按照《ANDA:评价经皮和局部给药系统的黏附性指南(草案)》(以下简称黏附性指南)开展黏附性研究,按照《ANDA:评价经皮和局部给药系统的刺激性和致敏可能性指南(草案)》(以下简称I/S 指南)开展I/S 研究,见表2。
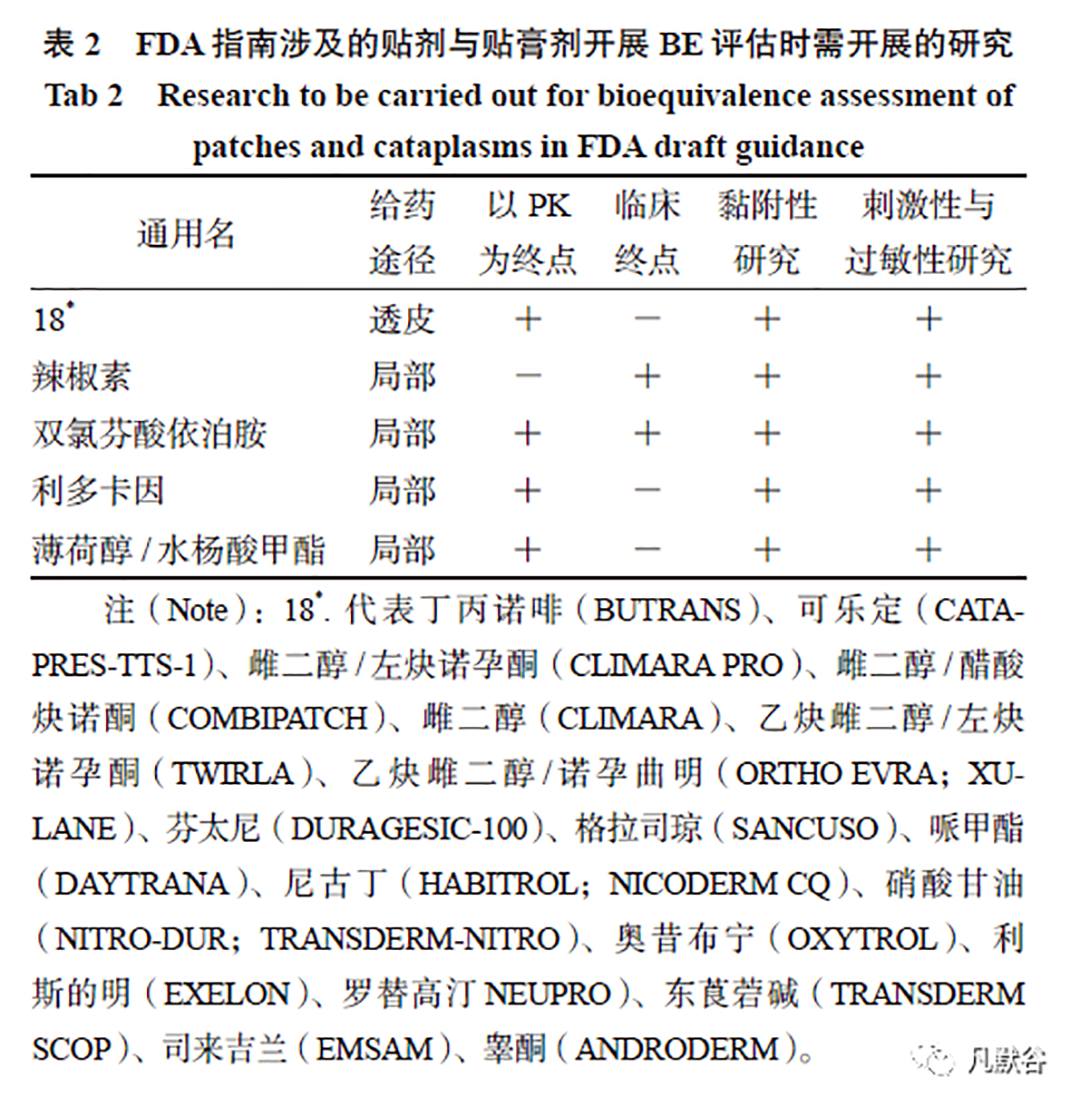
以PK-BE 研究
试验设计
研究设计为单剂量、两治疗、两周期、体内交叉。具体的设计和实施参考FDA《ANDA 提交的药物以药代动力学为终点评价指标的生物等效性研究》修订指南草案。在整个PK-BE 研究过程中,应监测并记录每个贴剂的黏附情况。
受试者的选择
受试者选择一般为健康男性及非孕、非哺乳期女性,部分制剂需根据其药物特点进行受试者选择,例如雌二醇的受试者选择没有雌激素治疗禁忌的绝经后妇女,雌二醇/ 左炔诺孕酮和雌二醇/ 醋酸炔诺酮选择没有雌激素治疗禁忌的不吸烟的绝经后妇女,尼古丁应选择健康吸烟者,睾酮选择睾酮缺乏(性腺机能减退型)的18 ~ 65岁的男性受试者。需要注意的是,此类制剂的PK-BE 研究需排除拟用药部位及对称部位存在肤色差异明显、毛发过多、疤痕组织、纹身、开放性溃疡、晒伤或身体穿孔等影响药物吸收的情况的受试者。此外,FDA 建议申请人也可根据具体研究附加受试者纳入标准和排除标准。
样本量
交叉设计的样本量需考虑的因素包括:① 检验水准α ,通常为双侧0.1(双单侧0.05);② 检验效能1 -β,通常至少为80%;③ 个体内变异系数(within-subject coefficient of variation, CVw%),可基于文献报道或预试验结果进行估计;④ 几何均值比(geometric mean ratio, GMR);⑤ 等效性界值[11]。
给药
药物规格
由于贴剂与贴膏剂的吸收机制不同于口服和静脉注射药物,对于规格的考量需要引起足够的重视。FDA 对透皮贴剂的规格通常用递送量/ 释放时间表示。FDA 已批准的透皮贴剂均以释药速率(mg·h - 1 或mg/24 h 等)作为规格,国内已上市透皮贴剂的规格表示有载药量、载药量/ 贴剂面积、递送速率等多种表达方式,透皮贴剂中载药量通常高于递送剂量,因此在选择参比制剂时需统一规格单位,避免因此造成的不等效。FDA 已批准的局部作用贴剂和贴膏的规格为基质中含有原料药的百分比,国内已上市的凝胶贴膏的规格为原料药含量及含膏量,局部贴剂多为原料药含量,与之相同。
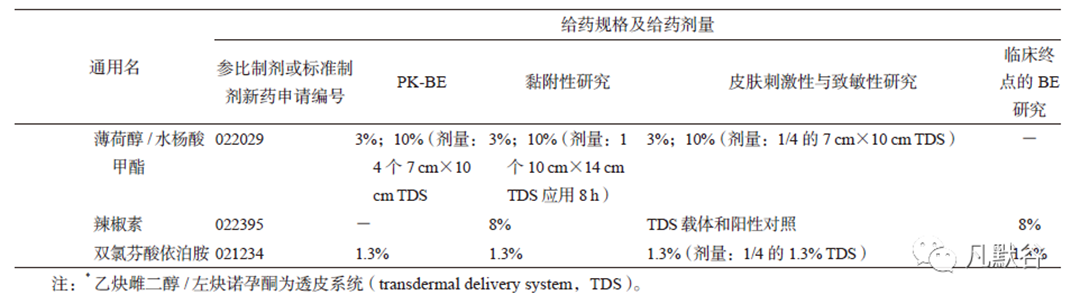
BE 指南对贴剂与贴膏剂PK-BE 研究中的剂量与规格都作了明确规定。薄荷醇/ 水杨酸甲酯的给药剂量为7 cm×10 cm(4 个TDS)或10 cm×14cm(2 个TDS),5% 利多卡因与1.8% 利多卡因的给药剂量均需3 个TDS,其他制剂的给药剂量均无特殊要求,具体给药规格及剂量见表3。
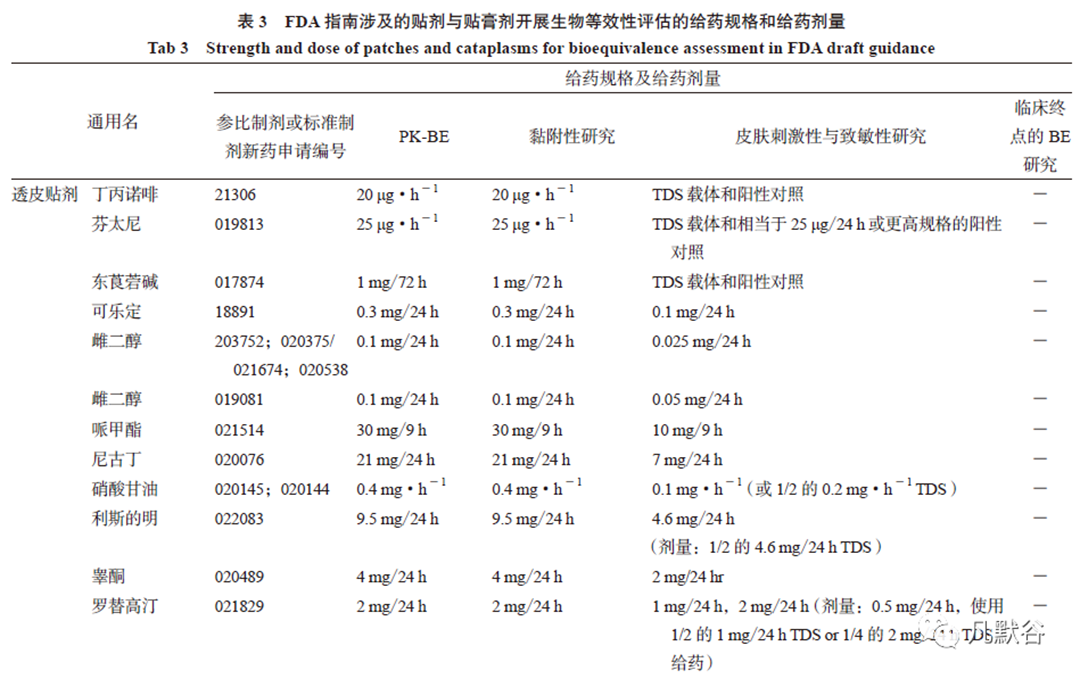
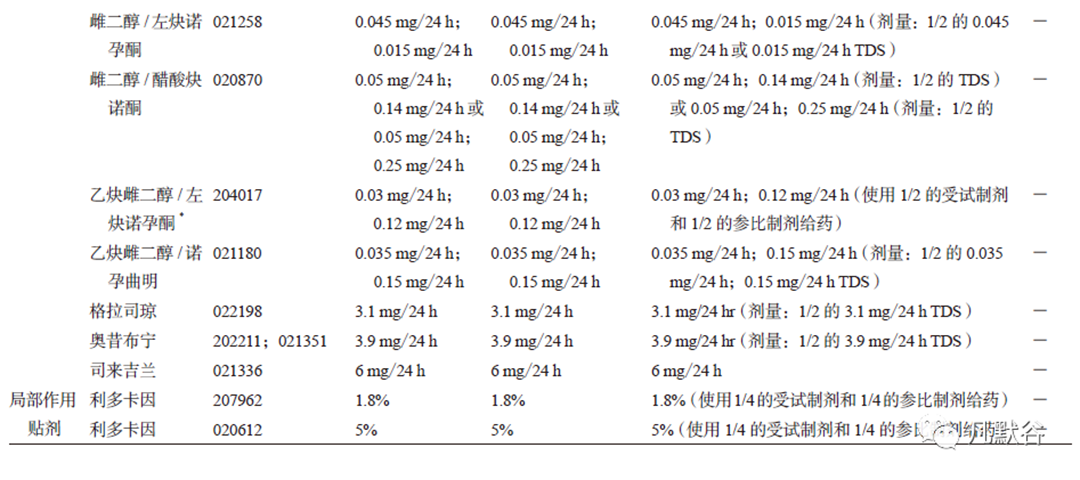
给药时间
不同制剂在PK-BE 研究中的给药时间有不同要求,丁丙诺啡、可乐定、雌二醇/左炔诺孕酮、乙炔雌二醇/ 左炔诺孕酮、乙炔雌二醇/ 诺孕曲明、格拉司琼佩戴7 d,雌二醇、雌二醇/ 醋酸炔诺酮佩戴84 h,芬太尼、东莨菪碱佩戴72 h、尼古丁、利斯的明、罗替高汀、司来吉兰、睾酮佩戴24 h,利多卡因、硝酸甘油、双氯芬酸依泊安佩戴12 h,薄荷醇/ 水杨酸甲酯佩戴8 h,哌甲酯佩戴9 h。
给药部位
给药部位的选择需参考参比制剂说明书中的用法,交叉试验中受试制剂与参比制剂的用药部位应为同一解剖部位的对侧。整个研究期间,避免按压或加固制剂,已脱离的制剂部分不可再次黏附于皮肤。
评价指标
与普通口服制剂的BE 评价指标相同,以Cmax、AUC0 ~ t、AUC0 ~ inf 为评价指标。
黏附性研究
贴剂与贴膏剂需按照黏附性指南开展黏附性研究。黏附性指南关注贴剂或贴膏剂在人体的黏附性的实际情况,当制剂失去对皮肤的黏附时,皮肤对经皮给药系统中药物的吸收发生改变,PK结果随之改变。另外,随着制剂完全脱离的可能性增加,药品对非计划用药人群的意外暴露风险同样增加,因此贴剂或贴膏剂的黏附性评估对于评价其安全性和有效性至关重要[12]。
试验设计
研究设计为单剂量、两治疗、两周期、体内交叉。黏附性研究可以单独开展,也可以与PK-BE 研究合并开展。
受试者的选择
受试者的选择与同一制剂开展PK-BE 研究受试者的选择标准基本一致。
样本量
黏附性试验为非劣效研究,样本量需考虑的因素包括:① 检验水准α ,通常为双侧0.1(双单侧0.05);② 检验效能1 -β ,通常至少为80%;③ 非劣界值为0.15。由于黏附性量表的离散性质,建议采用比标准假设计算出的值更大的样本量。PK-BE 研究与黏附性研究如果设计合并开展,需适当增加样本量,以保证PK-BE 终点与黏附性终点2 个独立的观察指标均有足够的把握度。
给药
黏附性研究的给药部位、给药规格及给药时间与PK-BE 研究方案一致,不同药品的黏附性研究的给药规格见表3。如有不同规格时,如安全性允许,尽量选择最大尺寸(或最高规格)的受试制剂进行黏附性试验。
黏附性研究评分标准
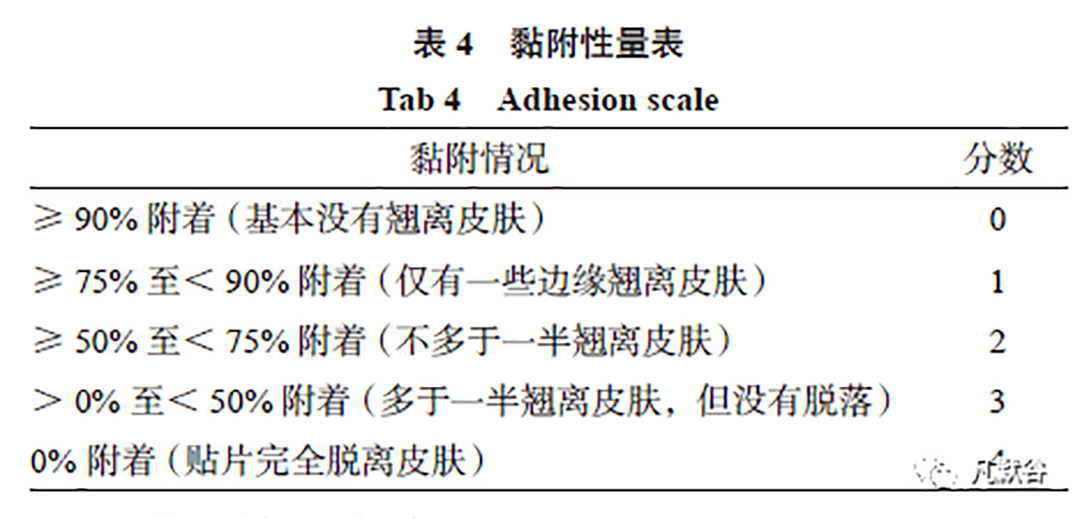
FDA《黏附性指南》提供了黏附性评分量表(0 ~ 4 分量表,见表4),每一个受试者在给药后的不同时间点观察制剂与皮肤的黏附情况并根据量表评分。
黏附性评估时间
为使受试制剂(T)和参比制剂(R)的黏附性在整个用药期内进行合理的比较,应在制剂应用后的多个时间点对T 组和R 组的每个制剂的黏附性进行评价。评估时间点的设计应等距分布,例如连续或累计使用7 d 的制剂应至少每日一次并在等距时间点评估一次;连续或累计使用72 h 的制剂应至少每12 h 评估一次;连续或累计使用介于12 h 和24 h 的制剂应至少每4 h 评估一次;连续或累计使用9 h 的制剂应至少1 h 评估一次。
评价标准或评价指标
主要终点指标
按黏附性评分系统进行评分后,计算在所有等距时间点(除外基线和时间0)内每个评估时间点取平均值得出的各个黏附性得分得出的平均黏附性得分(x =nΣi= 1 xi/n),T 与R 相比应呈统计学非劣效性,非劣界值为0.15。
次要终点指标
① 比较T 与R 在任何时间点黏附性得分大于2 的受试者的比例;② 计算并比较T 组平均黏附性得分大于相应R 组平均黏附性得分≥ 1 的受试者的比例与T 组平均黏附性得分大于相应T 组平均黏附性得分≥ 1 的受试者的比例;③ 比较T与R 从开始使用到黏附性评分≥ 2 的时间。
注意事项
① 黏附性评分需由经过培训的研究者,对每个时间点独立评分;② 黏附性研究与PK-BE研究合并开展时,黏附性研究的样本量可能大于PK-BE 研究的样本量,应在方案中预先规定PK-BE 研究的受试者的随机方法;③ 用药部位不可使用化妆品、面霜、乳液、粉或其他外用产品,用药前剪去多余毛发;④ 试验制剂的表面不可使用覆盖物或封盖物进行遮盖,防止按压或加固试验制剂,试验中已脱离的制剂不可重新黏附于皮肤;⑤ 整个研究期间,受试者的活动一般不应受到限制,用药时间大于24 h 不应限制受试者淋浴,但需避免剧烈活动;⑥ 在黏附性试验结束(或期间脱离)时取下的所有制剂应予以保留,进行残留药物含量分析。
皮肤刺激性和致敏性研究
贴剂与贴膏剂的配方或材质会阻止皮肤水蒸气传输的程度,或者由于其他因素也可能会对皮肤产生刺激或导致过敏反应。此类反应引起患者的不适,从而影响患者用药的依从性、皮肤渗透性以及制剂与皮肤的黏附性,最终可能会影响皮肤对制剂中药物的吸收,较严重的皮肤刺激还可能会影响制剂的安全性[13-14],因此贴剂与贴膏剂的刺激性与致敏性研究是其生物等效性评价不可缺少的一部分。刺激性研究可与致敏性研究合并开展,也可以单独开展。
试验设计
研究设计为随机、评估者盲、受试者体内重复。一般皮肤刺激性与致敏性研究设计为T 与R的对比研究,如由于有效成分的安全性问题,也可设计为TDS 载体与阳性对照研究,如辣椒素、丁丙诺菲。整个研究期间需同时进行黏附性评估。
受试者选择
I/S 指南中强调了I/S 研究中受试者的排除标准: ① 怀孕或哺乳期受试者;②有重大皮肤病或病症的病史,如过敏、牛皮癣、白癜风,或已知改变皮肤外观或生理反应的病症(如糖尿病或卟啉病);③ 有显著影响免疫反应的疾病病史[ 原发性或获得性免疫缺陷(如HIV 或AIDS),过敏性疾病(如过敏反应、哮喘或全身药物反应),肿瘤(如淋巴瘤或白血病),类风湿关节炎];④有皮肤癌病史(如黑色素瘤或鳞状细胞癌),基底细胞癌除外,基底细胞癌为浅表性且不涉及经皮给药系统应用部位;⑤ 试验前3 周内使用过显著影响试验制剂反应或改变对试验制剂的炎症或免疫反应的药物或治疗(例如环孢素、他克莫司、全身或局部皮质类固醇、细胞毒性药物、免疫球蛋白、卡介苗、单克隆抗体或放射疗法);⑤试验开始后72 h 内在用药部位使用抗组胺药或外用药物。
样本量
I/S 试验为非劣效试验,样本量需考虑的因素包括:① 检验水准α ,通常为双侧0.1(双单侧0.05);② 检验效能1 -β ,通常至少为80%;③非劣界值为0.20。致敏性试验单独开展或其与刺激性试验合并开展,需要至少200 例受试者。
给药
透皮贴剂
I/S 研究为个体内重复试验,在连续给药周期内,受试者需在身体对称部位同时应用T 和R,因此选择合适的规格和剂量,对研究的可实施及受试者的安全至关重要。不同药物制剂,BE 指南中明确了I/S 研究的给药规格与给药剂量。雌二醇/ 左炔诺孕酮、雌二醇/ 醋酸炔诺酮、乙炔雌二醇/ 左炔诺孕酮、乙炔雌二醇/诺孕曲明、格拉司琼、奥昔布宁以及司来吉兰的给药规格同PK-BE 和黏附性研究;可乐定、雌二醇、哌甲酯、尼古丁、硝酸甘油、利斯的明、罗替高汀以及睾酮的给药规格小于PK-BE 和黏附性研究;丁丙诺啡、芬太尼及东莨菪碱需使用TDS载体和阳性对照来进行I/ S 评价。另外中利斯的明、雌二醇/ 左炔诺孕酮、雌二醇/ 醋酸炔诺酮、乙炔雌二醇/ 左炔诺孕酮、格拉司琼、奥布西宁的I/S 研究给药规格为PK-BE 和黏附性研究的1/2TDS,利多卡因、薄荷醇/ 水杨酸甲酯、双氯芬酸依泊安I/S 研究的给药规格为PK-BE 和黏附性研究给药规格的1/4 TDS,不同制剂的具体给药规格见表3。
贴膏
利多卡因与薄荷醇/ 水杨酸甲酯的I/S 研究的给药规格与PK-BE 及黏附性研究相同,给药剂量与其他两个研究相比较小,为规格的1/4TDS。
局部贴剂
由于安全性考虑,辣椒素的I/S研究需使用TDS 载体和阳性对照进行评估;双氯芬酸依泊安在PK-BE 研究、黏附性研究、临床终点的BE 研究及I/S 研究中的规格一致,但I/S 研究的给药剂量小于其他研究,为规格的1/4 TDS。
评分系统
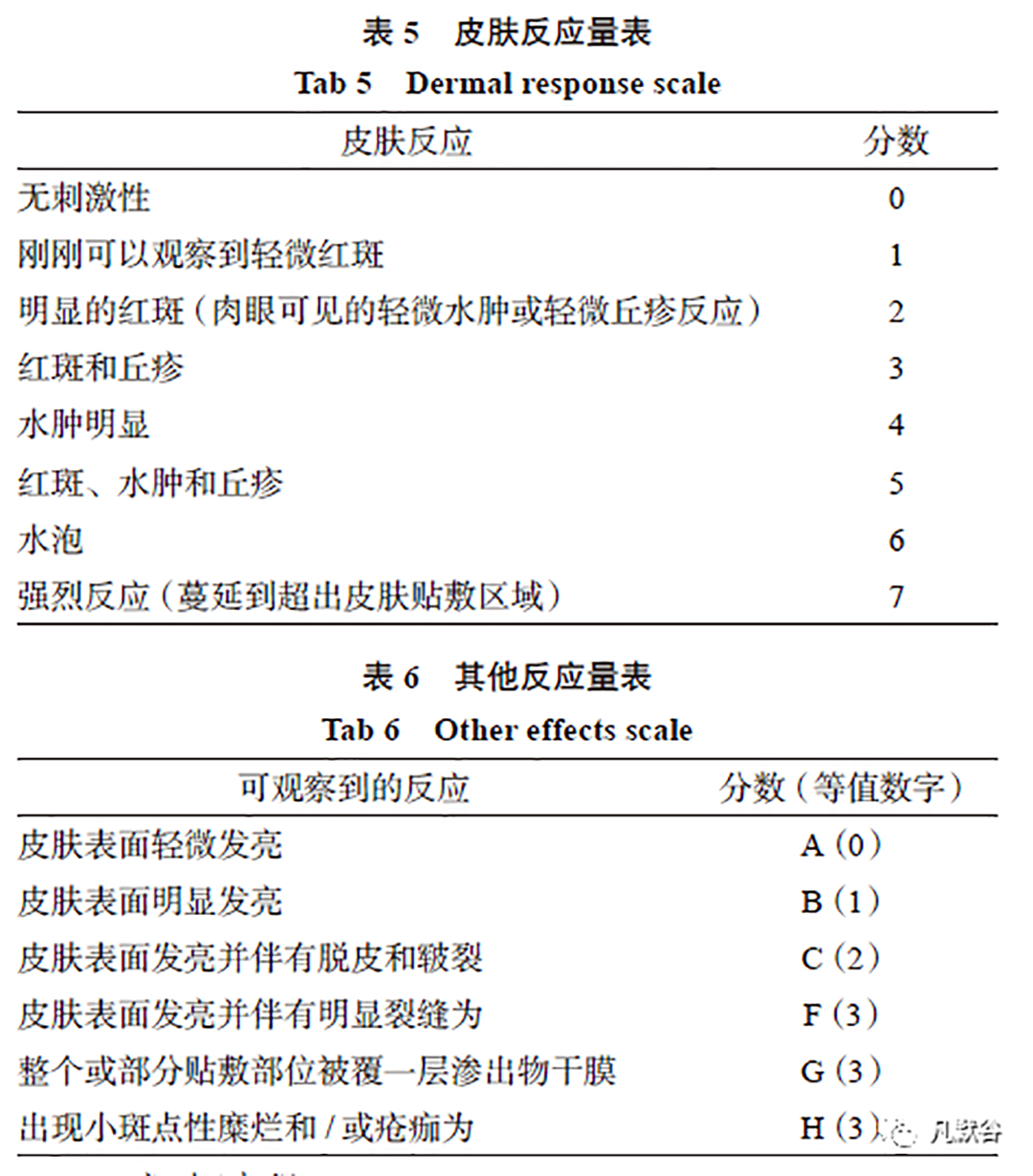
I/S 指南中给出皮肤反应量表与其他反应量表,研究中需记录受试者皮肤反应分数、其他反应分数以及总分(见表5 和表6)。
试验过程
致敏性研究的试验过程可分为21 d 诱导试验,14 ~ 17 d 休息期,48 h 激发试验。皮肤刺激性研究在21 d 诱导阶段完成。
诱导试验
每位受试者同时使用T 和R,连续使用21 d。用药时间和用药部位参考R 制剂说明书的用法用量进行设定,T 和R 应用于同一解剖部位的对侧。试验实施中,由经过培训的研究者对每个时间点独立评分。如果用药部位出现严重的皮肤刺激反应,应停止在该部位使用受试贴剂,可将制剂应用于新的部位。
激发试验
每位受试者同时使用T 和R,连续使用48 h。激发试验的用药部位需为诱导试验没有使用过药物的新的皮肤部位。在移除制剂后的30 min、24 h、48 h 和72 h,由研究者使用空白量表独立评分,并记录所有皮肤反应,研究者根据受试者发生的皮肤反应判定是否为致敏反应。对于所有表现出致敏反应的受试者,应在激发试验的4 ~ 8 周后再次进行激发试验。
评价标准
皮肤刺激性试验
刺激性试验评价的主要终点为①计算每贴制剂的平均累计刺激性积分和总刺激性累计积分,② T 与R 相比应呈统计学非劣效性[ 非劣效性(NI)边际:0.2];次要终点为①产生过度刺激反应的受试者比例,② T 组产生过度刺激反应的受试者比例不应高于R 组,③在用药期间,T 组产生的刺激反应不应早于R 组。
致敏性试验
除了对诱导试验进行上述相同评估外,还需在激发试验的评估时间点评估受试者的皮肤反应。致敏性的判定需满足以下所有标准:① 受试者至少有一个评估时间点出现在激发期制剂移除后24 h 以上(例如48 或72 h);②受试者在激发期的最后一次评估中的综合刺激评分至少为2 分;③如果受试者完成了两次激发期试验,两次试验结果需都满足上述两条标准。
试验豁免
对于已知为皮肤致敏剂的贴剂或贴膏剂(例如哌醋甲酯透皮贴剂),如从有效成分和制剂组成方面可证明T 的致敏性低于R,可考虑不进行致敏性研究。
注意事项
① 整个研究期间,受试者应避免用药部位暴露于加热垫、电热毯、加热灯、桑拿浴室、热水浴缸、热水床以及长时间的阳光直射等外部热源;
② I/S 研究可考虑使用胶带或其他覆盖物固定试验制剂,因胶带或其他覆盖物产生的皮肤刺激反应需单独记录;
③ 如试验制剂完全脱落,受试者需在24 h内更换新的制剂继续试验,试验制剂脱落超过24h 未能更换制剂的受试者应按照排除脱落处理。
讨论
近年,国内已上市的贴剂仿制药产品40 多个,近30 个为透皮贴剂产品。由我国自主研发的二类新药利多卡因、洛索洛芬钠、酮洛芬等凝胶贴膏陆续上市,国内对于贴剂及贴膏剂的研发热度不断升温。
国内还没有发布具体关于透皮贴剂、贴膏剂以及局部作用贴剂的BE 研究的指导原则,已备案的在研贴剂与贴膏剂仿制药的BE 研究仅部分参考了国外相关指南,具体试验设计是否能够合理且全面地评估其有效性和安全性仍存在疑问,贴剂与贴膏剂仿制药BE 研究的科学性及合理性仍需关注和完善。
另外,日本和南非认同的贴剂BE 研究还包括皮肤药代动力学(DPK)方法,由于角质层的药物浓度并不足以与疗效建立固定联系,DPK 指南于2002 年被废除[14]。笔者认为,新的医药用高分子材料的出现为透皮贴剂的研发提供了物质基础,加上检测手段的提高及药动学的发展,可以确定体内外的相关性,一些曾经采用DPK 方法研究的药物。
另外,在FDA 的指南中,所有评分需采取独立盲法评估,即由经过培训、处于盲态的研究者对每个时间点独立评分。指南中提到PK-BE 及黏附性研究中贴剂在使用过程中不能按压、加固、遮盖制剂,试验中已脱离的制剂不可重新黏附于皮肤;而I/S 研究则可以加固制剂,需特别注意。在全球新药研发失败率越来越高,开发新靶点越来越难的情况下,改良型新药具有成功率高、回报高、风险低、生命周期长等特点,已成为全球新药研发最佳选择。透皮给药系统在改良型新药的开发中有其特有的临床优势,且显示了较好的市场价值,本文也为改良型透皮贴剂的开发及临床研究提供了思路。
中国已经成为了ICH 成员国和ICH 管理委员会委员,国家药品监督管理局在对标国际标准,大力推进国内指导原则体系建设的同时,企业需根据制剂本身有效性与安全性的要求,并参考国外相关指导原则要求开展贴剂与贴膏剂的临床研究。
来源:《中南药学》2022 年
作者:李艳,李晓洁,王秀英,汤博凯,安雅翾,刘亚利
北京科林臻和医药科技有限公司
汕头大学医学院第一附属医院
本网站刊载的各类文章重在分享,尊重原创,如有侵权请联系我们(it@haigetang.com),我们将会在第一时间之内删除。


